
【研究背景】
在全固态锂电池(All-Solid-State Lithium Batteries,ASSLBs)领域,实现高能量密度与高安全性是当前产业界与学术界的共同目标。相比传统有机电解液体系,固态电解质在提高电池安全性、防止副反应、提升循环寿命方面具有显著优势。然而,尽管近年来出现了诸如硫化物、氧化物等各类固态电解质与正极材料组合,实际的能量密度提升却仍面临瓶颈。尤其是传统氧化物正极材料普遍存在容量低、离子电导率不足及界面兼容性欠佳的问题,导致全固态电池在实际应用中难以达到预期的高比能要求。
在这一背景下,研究者们开始探索新型正极材料体系,试图打破传统单电子转移机理的限制。多电子转移反应(multi-electron transfer)被认为是提升正极材料能量密度的重要路径之一。然而,实现多电子转移需满足特定结构与离子传导条件,而这在传统氧化物材料中并不容易。
针对这一难题,研究团队聚焦于卤化物(halide)基正极材料,提出利用Li2FeClx(X=Cl, Br)家族材料,实现三电子转移的嵌入-转化(intercalation-conversion)反应机制。这类材料不仅离子电导率高,而且能够在充放电过程中灵活实现从嵌入到转化的多阶段反应,使得正极材料展现出比传统氧化物更高的可逆容量与更优能量密度。
该研究的创新点在于:通过合理设计卤化物晶格结构与多电子转移途径,研究者将全固态电池的容量与能量密度提升到一个新的高度,为后续高能量、长寿命全固态电池的产业化提供了重要的技术储备。同时,这一工作也为未来开发其他多电子转移的正极体系提供了全新思路与可借鉴的范例。
【结果与讨论】
1、实验设计与材料制备方法概述研究团队采用高能球磨法(Ball-Milling)对LiCl和FeCl2(或FeBr2)以化学计量比例进行混合,并通过一系列固相反应,制得了Li2FeClx(X=Cl, Br)正极材料。由于论文重心在材料机理与性能展示,实验条件与设备描述相对简洁,但核心点在于:通过合理配比与工艺控制,获得具有高室温离子电导率与稳定结构的卤化物基正极材料。
论文强调离子导电性在全固态电池正极中的关键作用。与传统氧化物正极相比,该卤化物体系通过优化晶格通道与点缺陷结构,实现了更低的离子迁移能垒,从而在室温下(RT)即表现出高达3×10-5 S/cm的离子电导率。
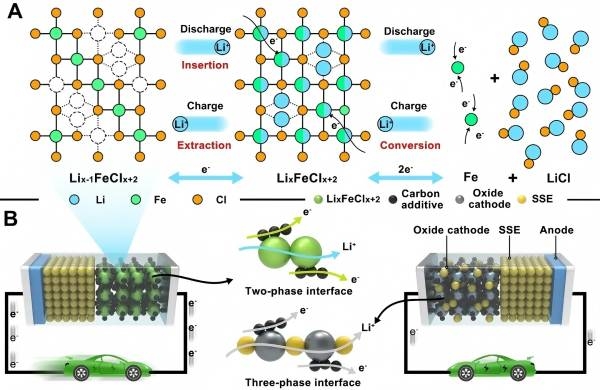
图1:(A) 基于插层转化机理的 LixFeClx+2 族反应过程示意图。(B) 使用 LixFeClx+2 族和氧化物正极在界面处对 ASSLB 进行比较。
2、材料结构与离子传输特性
关键数据与表征手段:
X射线衍射(XRD)结果表明Li2FeClx材料具备无序且近似正交晶格结构,铁元素占据晶格中的特定Wyckoff位置,而氯离子分布有序但在整体框架中为锂离子提供可移动通道。
扩散路径与迁移能垒分析(通过Ab Initio 分子动力学与BVSE分析)显示,Li离子在该晶格中拥有多条低能垒迁移路径,典型的能垒值约为0.48 eV甚至更低,显著优于传统氧化物材料中常见的≥0.5~0.6 eV的能垒。这意味着在室温下,Li2FeClx正极中Li离子将更为灵活地进行扩散。
重要参数强调:
室温离子电导率约为3×10-5 S/cm:与部分高性能硫化物电解质相比虽然仍有差距,但对于正极材料而言已是极大提升。
能垒值:0.48 eV的迁移能垒对比传统材料往往低数十个毫电子伏特,这种差异将在长循环过程中降低极化,提高可逆性。
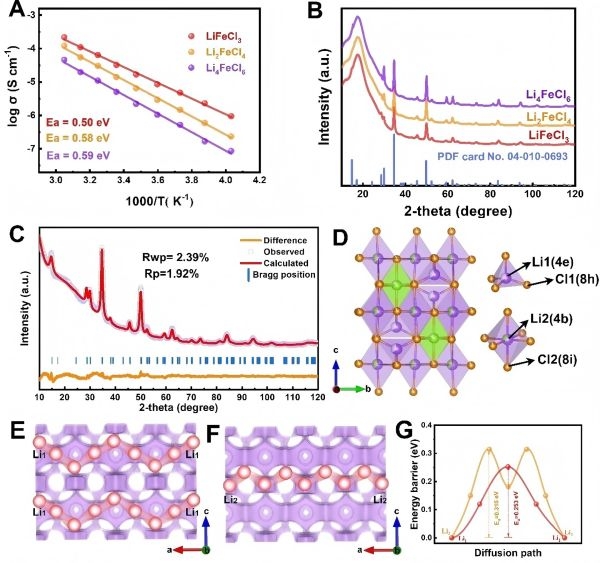
图2:LixFeClx+2的结构和扩散特性。
3、电化学性能表现:多电子转移与高容量实现在全固态电池组装中,研究者使用该Li2FeClx作为正极活性材料,搭配高离子电导率固态电解质及Li金属负极,组装全固态软包电池,并通过恒流充放电、循环伏安(CV)、交流阻抗(EIS)等手段表征其电化学性能。
关键实验结果:
放电容量与电压平台:与传统氧化物材料中单电子转移相比,Li2FeClx体系的多电子转移机制可实现高达446 mAh/g的放电比容量。这是一个极具冲击力的数据,相比市面上典型的正极材料(如LiCoO2等)的140-200 mAh/g容量有大幅提升。
能量密度:结合高容量与合适的操作电压区间,该材料的能量密度可高达912 Wh/kg,在全固态条件下这是一个非常亮眼的指标。
界面与结构稳定性:
论文中强调了该正极材料在充放电过程中经历从“嵌入”(Li离子从晶格中可逆抽取/插入)到“转化”(Fe-Cl键被打破,形成LiCl及金属Fe相)的反应阶段性转变。与传统碱金属氯化物体系易在循环中发生不可逆相变不同,这里的嵌入-转化反应在电压回程过程中可逆恢复,展现出结构重组的可逆性。这得益于:
特定晶格排列提供了稳定的离子通道与结构缓冲。
金属Fe纳米相与LiCl生成后,在低电压下重新转化回Li_2FeCl_x,使体系恢复初始结构,实现重复循环。
创新点:
三电子传输过程:这是论文的最大亮点。当一般正极材料仅通过Fe3+/Fe2+单电子转移来获得容量,该体系则利用Fe从Fe3+到Fe0的多级价态变化,贡献多电子数目,从而显著提升容量与能量密度。
双相界面到三相界面设计:论文指出,在合适的固态电解质与碳添加剂引入下,体系从双相界面(正极材料/电解质)过渡到三相界面(正极材料/电解质/导电炭黑),有利于进一步改善界面反应动力学与电子导通,确保多电子反应路径的高效进行。
在长循环条件下,具有高初始容量并不稀奇,但若能在数百次乃至上千次循环中保持稳定,就体现出材料的实用价值。文中展示的循环数据表明:
在较低的电流密度下(如0.1 mA/cm2),电池在多轮循环后仍能保持稳定容量。
容量保持率与库仑效率维持在较高水平,表明多电子转化与结构重组过程并未在长循环中导致不可逆相变或副反应累积。
由此可见,该卤化物正极具备优良的循环稳定性与高可逆性。这对于全固态电池的产业化至关重要。
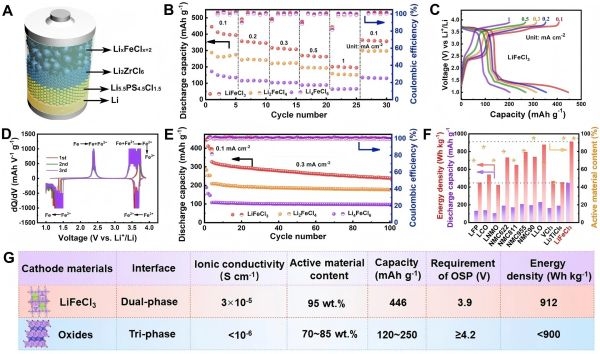
图3:LixFeClx+2的电化学性能。
5、界面相与电子结构分析研究者使用一系列先进表征手段(如原位XRD、球差校正TEM、X射线吸收光谱(XAS)、莫斯堡尔谱(57Fe Mössbauer)等)深入解析了嵌入-转化反应过程中Fe元素价态变化与局部结构演变。
重要发现:
利用Fe K-edge XANES与EXAFS分析,研究团队确认Fe元素在充电过程中从Fe2+逐渐上升到更高价态(类似Fe3+),而在放电时最终转化为近似Fe0状态的金属Fe纳米相。
随循环进行,这些金属Fe纳米相再与LiCl重新反应,回归原有的Li2FeClx晶格状态。这种完全可逆的相变为多电子传输反应奠定了基础。
57Fe Mössbauer谱证实了Fe环境的对称性与价态变化过程,与XAS结果相互印证。
通过这些高级表征,论文中呈现出一个清晰的反应机制图景:
初始状态下,Li2FeClx结构稳定,Fe处于+2价态。
充电(脱锂)过程中,结构逐渐释放更多Li离子,Fe价态上升,并最终形成Fe-Cl键弱化乃至被破坏,从而转化出Fe3+甚至Fe0物种。
放电(嵌锂)过程中,金属Fe再次与LiCl反应,重构回Li2FeClx结构,Fe回到较低价态。这一过程被称为可逆的嵌入-转化反应机制。
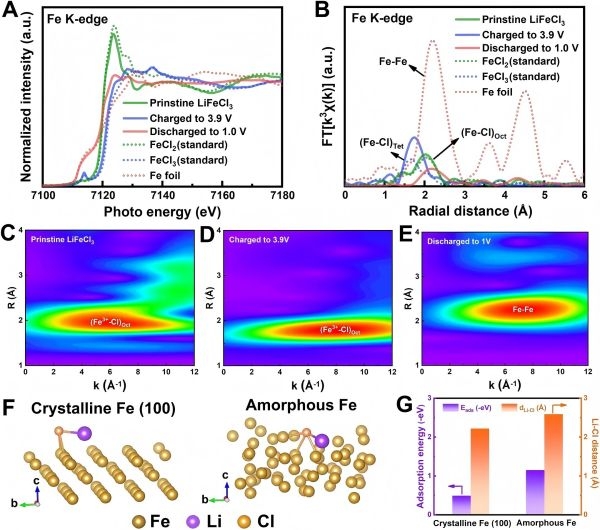
图5:LiFeCl3正极材料的充电/放电过程。图6:LiFeBr3正极材料的普遍性验证。6、对行业与科学问题的突破
传统全固态正极材料面临的行业痛点在于:
- 单电子转移导致容量有限,能量密度不高。
- 离子电导率低,限制倍率性能与实际应用。
- 界面不稳定导致循环寿命不佳。
本研究通过多电子转移机理与卤化物晶格设计,成功实现:
高容量(446 mAh/g)与高能量密度(912 Wh/kg),填补了传统氧化物正极在能量密度提升上的空白。
高离子电导率与低迁移能垒,加强了离子传输效率,可在较低温度下实现快速循环。
高循环稳定性,可逆的嵌入-转化过程确保电池多次充放电仍保持较高活性物质利用率。
同时,研究对行业应用前景的指引在于:
将来可针对Li2FeClx类卤化物正极进一步优化,如更换Fe以外的过渡金属、调控晶格参数或局部缺陷,持续提升离子导电与可逆容量。
与高离子电导的固态电解质搭配,可加速全固态电池的产业化落地进程。
【总结】
该论文通过系统研究Li2FeClx(X=Cl, Br)卤化物正极材料在全固态锂电池中的行为,为实现高能量密度和高循环稳定性的全固态电池提供了新路径。研究发现:
多电子转移反应机制:Li2FeClx正极实现了从Fe2+到Fe0的多电子传递过程,显著提升了单次嵌脱锂反应可获容量,从而获得446 mAh/g的高比容量与912 Wh/kg的高能量密度。
离子传导与结构特性:该材料在室温下具备3×10-5 S/cm的离子电导率,低离子迁移能垒(约0.48 eV)确保锂离子在正极内迅速传输,为快速充放电与高倍率性能打下基础。
可逆性与稳定性:借助原位XRD、XAS和Mössbauer谱等表征手段,研究揭示了Fe在充放电中的价态与局域环境可逆变化,实现了从“嵌入”到“转化”的反应重复往返。这种结构与价态的可逆调控使正极材料在循环中维持高容量与稳定结构。
总体而言,本研究在全固态锂电池正极设计中开创性地证明了卤化物家族具备通过多电子转移来提升能量密度与循环寿命的巨大潜能。未来针对Li2FeClx体系的进一步改进与规模化测试,将有望推动全固态高能电池的实际落地,为电动汽车与储能领域提供更安全、更高效的能源解决方案。
【文献信息】
| 标题 | Multi-Electron Transfer Halide Cathode Materials Based on Intercalation-Conversion Reaction Towards All-Solid-State Lithium Batteries. |
| 网址 | https://doi.org/10.1002/ange.202416635 |
| DOI | 10.1002/ange.202416635 |
| 其他 | 期刊:Angew. Chem. Int. Ed. 作者:X. H. Zhou; Ming Jiang; Yuhao Duan; Zhenghao Jia; Cheng Yuan; et al 出版日期:2024-12-03 |


发表评论 取消回复